

Child Health
Dr Shan Narayanan
Thalassemia is a blood disorder. It is an autosomal recessive disorder. In this disorder, the inherited abnormal genes affect the production of haemoglobin. Haemoglobin is the protein in red blood cells that carries oxygen.
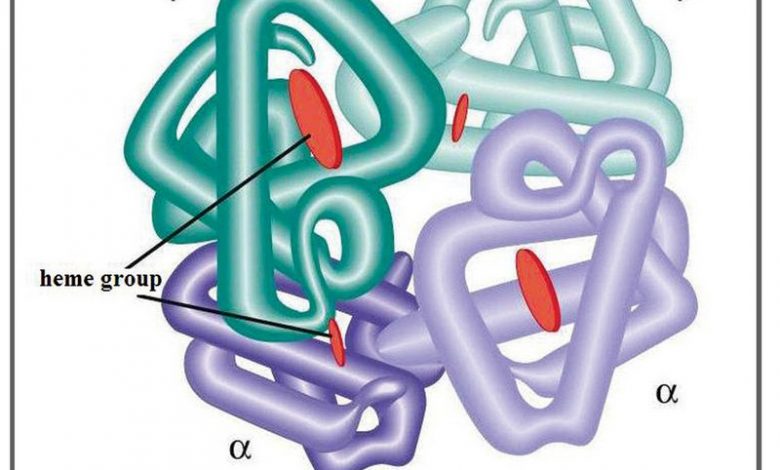
Haemoglobin is composed of four polypeptide (protein) chains, which in adults, consists of two alpha (a) globin chains and two beta (b) globin chains.
Individuals with thalassaemia make less haemoglobin and thus they have less red blood cell cells…they thus have anaemia. There are 2 types of thalassaemia, alpha thalassaemia (less alpha chains) and beta thalassaemia (less beta chains).
Four genes (two from each parent) are needed to make enough alpha globin protein chains. Alpha thalassaemia trait occurs if one or two of the four genes are missing. If more than two genes are missing, moderate to severe anaemia occurs.
The most severe form of alpha thalassaemia is called alpha thalassaemia major or hydrops fetalis. Babies who have this disorder usually die before or shortly after birth.
Two genes (one from each parent) are needed to make enough beta globin protein chains. Beta thalassaemia occurs if both genes are altered.
The severity of beta thalassaemia depends on how much both genes are affected. The defect results in moderate to severe anaemia. The severe form of beta thalassaemia is known as beta thalassaemia major.
 Thalassaemia affects males and females. The disorders occur most often among people of Italian, Greek, Middle Eastern, Southern Asian, and African descent. Severe forms usually are diagnosed in early childhood and are lifelong conditions.
Thalassaemia affects males and females. The disorders occur most often among people of Italian, Greek, Middle Eastern, Southern Asian, and African descent. Severe forms usually are diagnosed in early childhood and are lifelong conditions.
Thalassaemia is diagnosed from blood tests. Family genetic studies also can help diagnose the disorder.
If a mother expecting a baby and her husband are thalassaemia carriers, they can determine if the baby is affected through prenatal testing. Prenatal testing involves taking a sample of amniotic fluid or tissue from the placenta. Genetic testing is done on the fluid or tissue to make the diagnosis.
Thalassaemia makes the bone marrow expand, thus bones widen. This can result in abnormal bone structure, especially in the face and skull. Bone marrow expansion also makes bones thin and brittle, increasing the risk of broken bones. Their spleen is enlarged as they have to clear a lot of red blood cells.
Children with thalassaemia may have slow growth and delayed puberty. Treatment for severe thalassaemia often involves regular blood transfusions and folate supplements.
The regular blood transfusion gives rise to iron overload. As a result of iron overload, the individual may have many complications. They thus have to go on chelation therapy to remove excess iron from the body.
Bone marrow transplantation may help treat the disease in some patients. Bone marrow transplantation is most successful when the donor matches fully the patient’s special blood characteristics known as HLA antigens. This happens when very close relatives (sisters and brothers) offer their bone marrow for transplantation and when the patient is fairly young and well treated.
